



GPMAW - 生物蛋白质分析软件
GPMAW是一种详细分析蛋白质和多肽一级结构的程序。虽然重点是质谱分析,但您也会看到大量的物理/化学数据。
GPMAW程序主要用作蛋白质和肽的质谱分析工具,然而,很多其他生物信息学工具已被包括在内,因此该程序的使用远远超出了简单的质量分析。
该程序可在自Windows 2000以来的全部32位版本的Windows上运行(即2000、XP、Vista、Win7)。它还将当前的64位版本上运行,但尚未在此版本上全部测试。它还可以在带有Windows模拟器的Mac系统上运行,但不能保证全部兼容。
序列处理:通过在Entrez和本地数据库(FastA格式和Swiss-Prot)中直接进行数据库搜索,从多种不同格式导入序列。序列可以保存在本地文件(数据库)中以备将来参考。
从序列窗口可以执行大量操作。序列可以以FastA格式导出(单个或全部序列一次),以便轻松传输到其他程序。
质量分析:可以通过自动方法(例如定义酶作用的灵活命名法)或手动切割蛋白质。肽显示有很多参数(各种质量值-mono、ave、charges-Bull&Breese指数、HPLC指数、pI、charge)并且可以进一步处理(例如cross-linked, new cleavage)。
可以在FastA格式的任意本地数据库上执行肽质量搜索。
生物信息学:可以显示大量图表,可以在本地数据库上执行BLAST搜索。使用Clustalw进行多重比对。
CustalW可执行文件现在是GPMAw包的一部分,但在以前的版本中,需要自己安装。
序列窗口
序列窗口是GPMAW的中心窗口,是序列的默认视图。从这里您可以调用大多数其他与序列相关的功能(通过菜单、工具栏或弹出菜单)。
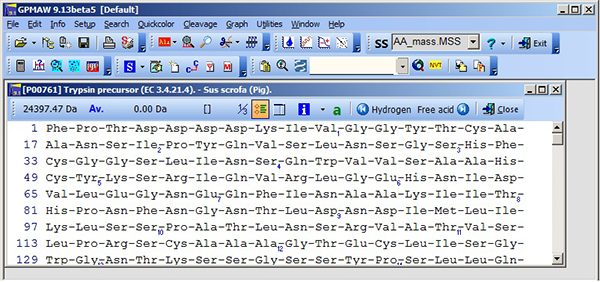
显示器可以多种方式配置:
1或3字母显示,
平均质量/单同位素质量,
固定残余宽度
具有序列信息的侧边栏,
显示二硫键、修饰残基等。
通过突出显示序列的一部分,您可以轻松获得给定肽的质量。为了便于导航,您可以为特定的残留物上色(有三种不同的颜色和下划线可用)。
大多数设置都可以在系统配置框中轻松预设。
序列窗口构成父窗口,可以从中创建大量派生子窗口:
肽窗口
Ms/Ms窗口
大规模搜索、合成搜索
图表:Hydrophobicity, secondary structure, charge vs. pH, dot-plot, alpha-helical wheel
肽窗口
肽窗口通常通过自动摘要命令从序列窗口调用。但是,还有其他方法,如手动或半自动切割。
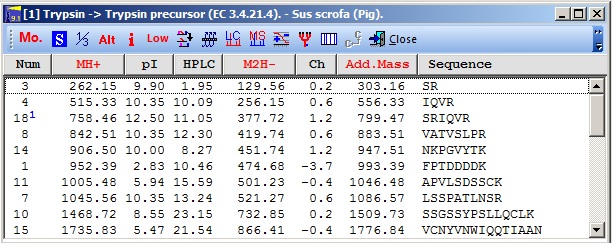
肽窗口列出了将由给定蛋白质生成的全部肽以及大量(>20)物理化学参数
电荷-单个/多个/负/正
肽数、
位置、
HPLC指数
theoretical pI
Bull & Breese index
序列-1/3字母等
可以通过部分分切割(图中蓝色上标表示)、修饰的末端、修饰的残基(甚至以有限的方式支持部分修饰)生成肽。为了在序列窗口中更方便引用而着色的特定残基被带到该窗口。显示的实际参数可以由用户配置。
通过点击标题可以对任意列进行排序。第二次单击会反转排序。
通过工具栏和/或弹出菜单(单击鼠标右键),您可以访问与消化(如模拟HPLC反相色谱图)或当前所选肽(ms/ms cleavage、肽信息、电荷与pH图)。
【英文介绍】
GPMAW is a program for the detailed analysis of the primary structure of proteins and peptides. While the focus is on mass spectrometricl analysis, you will also be presented with a large number of physical/chemical data.
GPMAW - General Protein/Mass Analysis for Windows
The GPMAW program is primarily intended as a tool for mass spectrometric analysis of proteins and peptides. However, a number of other bioinformatics tools have been included, so the use of the program extends far beyond simple mass analysis.
The program runs on all 32-bit versions of Windows since Windows 2000 (i.e. 2000, XP, Vista, Win7). It will also run on current 64-bit versions, but has not been thoroughly tested on this platforms. It can also run on Mac systems with a Windows emulator, but full compatibility is not guaranteed.
Except for the ms/ms search, the program does not need a strong processor or fast hard disk but can run on any system. Running the ms/ms search you need a screen with SVGA+ resolution, otherwise, you can even run it on a netbook.
Sequence handling: Import of sequences from a number of different formats with direct database search in Entrez and in local databases (FastA format and Swiss-Prot). Sequences can be saved in local files (databases) for future reference.
From the sequence window a large number of actions can be performed. Sequences can be exported in FastA format (either singly or all sequences at once) for easy transfer to other programs.
Mass analysis: The protein can be cleaved by automatic methods (e.g. a flexible nomenclature for defining enzyme actions) or manually. The peptides are displayed with a number of parameters (various mass values - mono, ave, charges - Bull&Breese index, HPLC index, pI, charge) and can be further worked upon (e.g. cross-linked, new cleavage).
Peptide mass searches can be performed on any local database in FastA format.
Bioinformatics: A number of graphs can be displayed, hydrophobicity, dot-plot, secondary structure prediction. BLAST searches can be performed on local databases. Multiple alignment using ClustalW.
The CustalW executable is now part of the GPMAw package, but on previous versions you have to install yourself.


- 2026-07-29
- 2026-07-28
- 2026-07-26
- 2026-07-24
- 2026-07-23
- 2026-07-21

- 2026-08-05
- 2026-07-30
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29











