



Sequencher - DNA序列分析软件
Sequencher是DNA 序列分析的工业标准软件。它可以和全部自动序列分析仪一同工作,并且因为它的Contig 组装、很短的学习曲线、用户友好的编辑工具,以及突出的技术而众所周知。从差不多15年前初次版本到现在,Sequencher在基因和制药公司中应用于序列分析任务,Sequencher被生命科学研究者们使用于很多不同的 DNA 序列分析应用方面,包括基因重组、突变检测、法医的人体辨别,以及分类学等等...
Sequencher 内含多种拼接算法,还有功能的序列编辑工具、具有ORF分析,酶切位点作图、杂合子识别等分析功能。可以应用于杂合子和SNP的检测和分析,cDNA到染色体DNA的大型间隙对准,比较排序,对置信评分的、ORD转换、GeneBank化导入、以及限制酶映射等等,序列比对可直接用MEGA操作,编辑更方便,比对时间稍短。Sequencher新版本为5.4.6。
DNA序列分析的新功能和增强功能:
Sanger:
-
从Primer-BLAST发送引物对序列,在Sequencher Connections中运行到Sequencher项目
-
使用项目窗口中的共有序列作为混合测序项目的NGS的参考序列
-
New Batch Revert Trim Ends命令
-
能够在项目窗口中调整字体大小
NGS:
-
更快的GSNAP 和 BWA-MEM工作流程
-
构建可重复用于与全基因组的GSNAP数据库和BWA索引
-
使用FastQC报告查看和保存NGS原始数据文件的质量得分和元数据
-
生成变体调用文件(VCF)以使用SAMtools标记NGS对齐中的变体
RNA-Seq:
-
扩展袖和套装,加入Cuffquant和Cuffnorm
-
Cuffdiff和Cuffnorm的条件和重复编辑器
-
增强的RNA-Seq可视化,具有自定义排序和过滤选项
-
使用增强的外部数据浏览器管理DNA-Seq和RNA-Seq项目
Sequencher 产品:
-
SNP 检测 (SNP Detection)
-
序列装配 (Sequence Assembly)
-
序列编辑 (Sequence Editing)
-
自动分析 (Automated Analysis)
-
矢量微调 (Vector Trimming)
SNP 检测
-
使用 Sequencher 来进行比较对准以便鉴别和报告 SNP 以及突变。
例如,调用峰,功能分析你的序列以寻找潜在的杂合子。你可以控制定义杂合子的严格度。 -
在装配总揽中表示出杂合子的位置
-
你可以从杂合子跳到下,而需要在 Base View 里面按一下空格键。你还可以观察及其参考序列的转换。
及其参考序列的区别会列在可导出的表格内。参考序列从装配到下装配时,SNP 的数目保持。
序列装配
Sequencher 的装配算法可以将你的 DNA 片断而又的装配起来。自觉的工具允许你在数秒内就可以设置好参数并且调整它们。
你可以不顾方向的装配序列。Sequencher 会比较前端和反转补体方向来装配可能的 Contig。
将 Sequencher 的多功能装配工具应用到:
-
确定矢量构造
-
装配病毒和细菌基因组
-
从 DNA 库中聚类数以万计的序列
-
将基因变体和参考序列做差异化数据
-
创建引物地图
-
将 DNA 装配成基因组序列
-
以及项目...
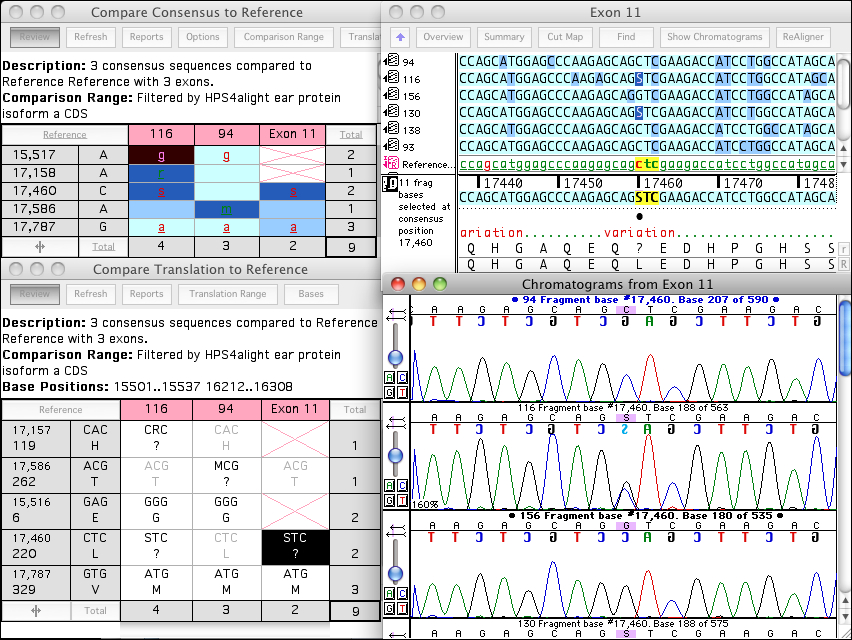
Sequencher能够给你足够的信心认为序列是决定正确的。你可以只观察序列的色谱图,你也可以从前端或反方向同时查看多个对齐的色谱图。在你的已对齐数据中滚动式很的。你可以使用Sequencher的选择工具来高亮不或者低质量的区域。
下一代DNA测序(NGS)
Sequencher通过将新的同行评审的NGS算法从命令行中带入直观的点击界面,为台式科学家提供。是执行参考引导的比对,从头组装,变体调用还是SNP分析,Sequencher都拥有获得结果的工具。Sequencher了的Cufflinks套件,用于深入转录分析和RNA-Seq数据的差异基因表达。Sequencher可以使用自定义图表和图表生成RNA-Seq数据的可视化,从而在几秒钟内为您提供可用于发布的图形。

Sanger DNA分析
Sequencher广泛的Sanger分析功能是它构建的基础。从头到尾可定制。凭借易于使用的界面已经磨炼超过25年,初次用户将在几分钟内感觉像人士。修剪读取,自定义装配和对齐算法,变化表,摘要报告,注释等内容。

序列编辑
Sequencher 能够给你足够的信心认为序列是正确的。你可以只观察序列的色谱图,你也可以从前端或反方向同时查看多个对齐的色谱图。 在你的已对齐数据中滚动是很的。你可以使用 Sequencher 的选择工具来高亮不或者低质量的区域。
自动分析
Sequencher 可对你的数据进行批处理,这个过程是透明、用户可定义、可恢复的,并且 Sequencher 不为了自动化的目的而破坏你科学结论的正确性。
当你工作在多个序列时,Sequencher 可以:
-
调用峰
-
调整矢量
-
调整低质量的尾端
-
创建序列
-
恢复到实验数据
Sequencher 总是管理两份数据,一份编辑过的,一份则是原始的导入数据。当你应用“Revert to Experimental Data”命令到在你项目中已选定的序列或者在一序列中的选中部分时,你可以撤销或者部分编辑动作。
自从 4.5 以后,Sequencher 就了新的自动化工具,“Assemble by Name”。依靠“Assemble by Name”,你可以选择片段名的一部分作为共享的标识符,或者说是“装配句柄”。Sequencher 就可以将选择和名字自动转换为 Contig。例如,通过按钮的点击,你就可以将 90 个文件,45对前端和反向序列转换成 45 个根据你的病人编号命名的 Contig。装配参数的变化都会重组你的片段,因此你可以根据克隆编号、日期、引物,或者你记录在序列名称中的,来装配 Contig。
如果你做了排序,你会感激“Assemble by Name”,尤其是在你有个带有标准引物集的样本序列时。事实上,在序列名中具有可用信息人的人,都有可能使用“Assemble by Name”。一些应用:
-
用于身份鉴定和人口研究的 MHC 和线粒体基因排序。
-
候选基因的 PCR 产品的分析。
-
对保存的跨系统分类物种基因的排序。
-
通过跟踪病毒对抗病毒剂或疫苗的抗药性,对病毒序列进行监控。
即使在项目中 Contig,“Assemble by Name”仍然是有用的,因为它 Contig 的名称可以较地表述样本的名称。如果你有命名为“Bob”的样本,那么正确命名你的 Contig 并不是什么难事,但你有象“Bob-2309888762-4321”这样更复杂的名字时,允许 Sequencher 命名你的 Contig 是有用的。
矢量调整
自动排序器以基础调用错误生成结果。从 DNA 库克隆的序列通常矢量序列、polyA 尾,或不相关序列。内含子和引物序列通常会和放大的外显子序列侧面相连。不调整的话,这些污染物会扭曲你的装配和下游分析。 Sequencher 允许你调整低质量或者含糊的数据。“Trim Ends”将令人误解的数据从序列片段的尾部去除。“Trim Vector”将污染序列尾部的序列数据移除。“Trim to Reference”剔除超出装配参考序列的序列尾部。
在执行调整之前,Sequencher 会显示计划调整的图形描述,这个功能允许你重新定义你的条件。
系统要求:
Mac要求:
Mac平台当前版本的Sequencher 5.4.6版本:10.7、10.8、10.9、10.10、10.11、10.12、10.13和10.14
在Mac上Sequencher的硬盘空间要求为355MB
在Mac上运行Sequencher的内存要求是8GB RAM。使用DNA-Seq工具处理NGS数据需要额外的6GN RAM。对于GSNAP,建议16GB。
Windows要求:
操作系统:Windows 10,Windows 8.1,Windows 8和Windows 7Sequencher 4.8和版本。Sequencher 5.4不再提供Windows XP。Sequencher 5.4.1不再提供Windows Vista。Sequencher 5.4.6不再Windows 7.
Windows上Sequencher的硬盘空间要求为280MB
在Windows上运行Sequencher的内存要求是3GB RAM
注意:Apple发行时,Sequencher5.4.6将与MacOS Catalina(10.15)不兼容。为了继续使用Sequencher 5.4.6,请暂时不要升级MacOS。


- 2026-07-29
- 2026-07-28
- 2026-07-26
- 2026-07-24
- 2026-07-23
- 2026-07-21

- 2026-08-05
- 2026-07-30
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29











