



Molcas - 量子化学软件
量子化学程序MOLCAS可以用量子化学模型研究分子体系,从SCF/DFT到耦合簇,从RASSCF到动态电子相关处理的MR-CI或MS-CASPT2。动态相关处理的多组态密度泛函理论(CAS-DFT)还在开发中。
MOLCAS的重点在于多组态的量子化学计算,用于研究单组态不能给出电子结构合理描述的体系,例如激发态,化学反应的过渡态,重元素体系(过渡金属,镧系,锕系),等。MOLCAS的另一是多组态的相对论处理(标量相对论和自旋-轨道耦合),并提供为相对论计算设计的基组。
MOLCAS可用于计算分子结构,键能,化学反应的能垒,激发能(自旋-轨道耦合),振动分辨吸收光谱,以及分子,等。MOLCAS可以用自洽反应场计算溶剂模型。新增加的QM/MM方法可用来计算大分子和分子簇,可以用半经验方法CNDO研究周期体系的能带。通过使用NEMO方法,MOLCAS还可以产生分子间作用力,用于MC/MD模拟。
MOLCAS并不是“黑箱”工具。要求用户应当受过量化方面的训练,对当今的一些量化模型有些了解。
OpenMOLCAS是遵守LGPL协议的开源MOLCAS代码部分,未经原作者授权的MOLCAS代码部分未在内。
目前Molcas的版本为8.6。Molcas是从头算量子化学软件包,基本原理是能够处理大多数元素周期表中的原子组成的分析的电子结构。这样,程序包的主要重点是多配置方法,其应用程序通常与高度退化状态的处理有关。
Molcas代码基于OpenMolcas的稳定版本。与Molcas附带和经过验证的发行版相反,OpenMolcas是开发人员的分支机构,其性通过自动测试进行验证。
Molcas的功能:
-
SCF / DFT , CASSCF / RASSCF , CASPT2 / RASPT2
-
代码
-
更新
-
Molcas不是黑匣子工具。用户应该是受过教育的量子化学家,对当今使用的不同量子化学模式,其应用领域和固有性有一定的了解。
在Molcas中实施的方法
-
跨多个进程运行可以使某些程序受益,这些程序用(p)表示,这并不一定意味着整个程序都可以地并行化。
-
代码
-
SEWARD:用于 和两个电子积分的程序,其中大量的积分(P)
-
MCKINLEY:解析积分二阶导数,与SCF和CASSCF一起使用以获得能量Hessians
-
AMFI:使用原子平均场近似生成自旋转轨道积分
波浪功能码
-
SCF:关闭并打开Shell SCF和DFT程序。(P)
-
RASSCF:RASSSCF(CASSCF)计算程序。(P)
-
MP2:二阶代码。(P)
-
CASPT2:具有CASSCF(RASSCF)参考函数的多配制二阶摄动理论。(P)
-
MRCL / ACPF:一种基于图形unit群形式主义的多参考CI程序。用于耦合系数。
-
CCSDT:布拉迪斯发套件的耦合簇代码。用于粉笔和受限制的高自旋开壳系统
-
CHCC / CHT3:基于布拉迪斯拉发的Cholesky分解套件,适用于大型封闭壳系统的耦合簇代码。(P)
属性和实用程序
-
MCLR:SCF / CASSCF线性响应程序
-
RASSI:计算RASSCF波函数之间的跃迁密度,并可以选择添加经能量校正的AMFI自旋轨道哈密顿量。
-
VIBROT:双原子分子振动波函数和跃迁的数值解
-
GENANO:构建密度矩阵平均ANO的程度
-
SLAPAF:执行几何并搜索通过渡状态和圆锥形交换
-
GEO:在内部坐标中执行约束的多篇段几何
-
MOTRA:一种用于单电子和二甸子积分的AO到MO转换的程序
-
GRID IT:用于在笛卡尔网格上计算密度和分析轨道的程序
-
SINGLE_ANISO:计算任意单核络合物和碎片的各向异性磁性能
-
EMBQ:计算点电荷的几何位置和值,它们在有限的体积中重现静电势
-
FALCON:基于片段法计算大型系统的总能量
-
DYNAMIX:对多种电子状态执行分子动力学(MD)模拟
环境影响
-
FFPT:用于有限场微扰计算的模块
-
PCM:使用可连续体模型添加反应场哈密顿量的模块
-
ESPF:结合了量子力学和分析力学计算
-
NEMO:单一的程序包,用于生成分子件力场的参数
-
QMSTAT:基于Metropolis蒙特卡洛模拟技术的混合QM/MM离散溶剂化模型
图形用户界面(GUI)
-
GV:基于OpenGL的GUI,用于可视化轨道的地编辑坐标文件
-
LUSCUS:基于GTK+GUI
应用示例
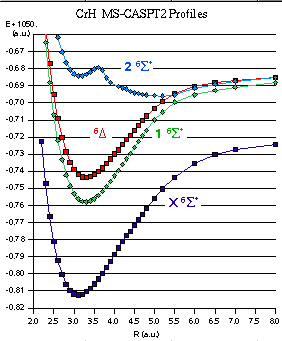
势能曲线
Molcas给出了势能曲线,可用于计算的分析,例如键矩和能量以及电子光谱
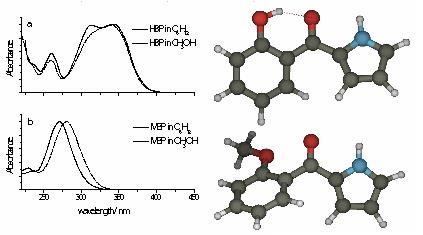
分子光谱
Molcas可以计算分析的电子吸收光谱,化合价,雷德堡和混合态
反应机理
Molcas允许研究化学领域的反应机理
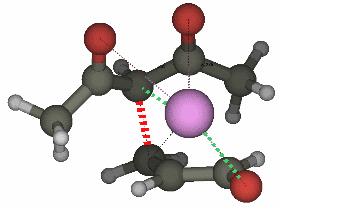
生物化学
Molcas及其与Tinker代码的接口允许生物系统进行QM / MM计算
在Linux下运行Molcas
注意,不建议使用某些版本的GNU编译器来编译为O3的Molcas。
另外,您可以使用商业编辑器,例如Intel , NAG , PGI , Absoft,PathScale , SunStudio。
在Mac OS上运行Molcas
Molcas现在也可以在MacOS下运行。但是,如果要使用xlf编译器,则在区分大小写的分区安装Molcas。
在MS Windows下运行Molcas
Molcas现在也可以在MS Windows下运行。您安装Cygwin。注意,出GNU gcc / g77外,不编译器。
波函,能量
-
MOLCAS使用以下波函模型进行总能量,电子结构和分子的计算:
-
Hartree-Fock以及和DFT组合。使用直接或者半直接方案,可以处理一千个以上的基函数。
-
DFT有:LSDA,LDA,SVWN,LSDA5,LDA5,SVWN5,HFB,HFS,BLYP,B3LYP,B3LYP5,TLYP,XPBE。
-
Moller-Plesset二级微扰理论(闭壳层或限制性开壳层),可用于计算动态电子相关影响。
-
MCSCF(CASSCF或RASSCF)用于处理电子结构无法用单行列式描述的体系。态平均方法可以处理多个电子态。可以研究一百万个电子组态的波函。
-
多参考二级微扰理论(CASPT2)可用于计算CASSCF电子态的动态电子相关能。可以用实能移动或虚能移动技术入侵态。6.2版还可以使用新的移动选项IPEAshift,它对开壳层体系可以排除关联能的系统误差,还可以入侵态。CASPT2的多重态版允许参考态用哈密顿方法进行相关能修正。
-
CASSCF/RASSCF波函可与基于DFT的相关势方法组合,获得动态电子相关的多组态波函(CAS/RAS-DFT)。这一选项将在MOLCAS-6以后的版本中发布。
-
对于小分子,可以用多参考CI(MR-CI,MR-CISD,RAS-CI和MR-ACPF)方法产生高精度波函和能量。
-
对于能够用单行列式很好描述的分子和原子团,可以用耦合簇方法(闭壳层和限制性开壳层的CCSD(T))研究。
-
变分价键程序CASVB。
-
能量和梯度可用于:Stoll-Dolg ECP基组的计算,全电子基组的标量相对论(二级Douglas-Kroll,Barysz-Snijders-Sadlej)计算,有限核近似。用Douglas-Kroll标量相对论修正,可以研究重金属原子(例如镧系和锕系)的体系。新的ANO-RCC基组可用于整个周期表元素的Douglas-Kroll标量相对论计算。
-
MCLR程序计算热力学,进行单个或两个同位素的取代计算。
-
.使用CNDO/INDO半经验哈密顿量进行3D能带计算。
-
用Pipek-Mezey方案产生局域化轨道。
分子结构,振动频率,热动力学
-
使用解析梯度技术的自动几何,可用于HF/DFT和RASSCF(RASDFT)波函。CASPT2结构可以用数值梯度程序获得。这些程序可用于获得基态和激发态的平衡结构,过渡态等。
-
通过解析二阶导数,对RASSCF波函计算振动频率和热动力学量。
-
SLAPAF程序可用于寻找能量路径和内反应坐标,以及交叉点的能量交点。
激发态和电子光谱
MOLCAS为研究激发态势能曲面而设计:
-
能量可以用的波函方法获得。几何也可以用于态平均RASSCF能量。
-
在RASSCF,用RASSCF态相互作用方法计算跃迁。这是MOLCAS程序所的。这个代码还可使用单电子SO哈密顿量和所谓的原子平均场积分(AMFI),计算自旋-轨道耦合。增强版本的RASSI-SO通过计算振荡强度和爱因斯坦系数A获得RASSCF理论的荧光和磷光寿命。
-
在RASSCF(RASDFT)的激发态势能曲面上自动搜索能垒,圆锥交叉点等。
-
用MULA代码计算两个电子态谐振能级之间的跃迁偶矩和振动跃迁的强度,获得振动分辨电子光谱。
环境影响
MOLCAS-6可处理溶剂中的分子和大分子体系:
-
用Onsager球穴模型或连续介质模型(PCM)处理溶剂影响。
-
QM/MM方法可用于计算大分子体系,如蛋白质,分子簇等。ComQum代码可以把Molcas和Amber分子力学代码(需另外获得)组合进行QM/MM计算。
-
NEMO程序产生分子间作用力,用于MC/MD模拟。这些力场静电,感应,色散和交换-排斥项。它们基于个别分子的计算。
功能
-
调用COLUMBUS的接口进行MR-CI(+Q),MR-ACPF, MR-CEPA, MR-AQCC(-v)等计算。
-
平台的图形用户界面,用于显示轨道和密度(尚在开发中)。
-
部分模块实现了并行化:Seward,SCF,Alaska,McKinley,MCLR和CC基本实现了线性或超线性加速。
-
结合了到ACES II的界面,可以和该代码的耦合簇一起使用。同时也可在ACES II中使用MOLCAS中的积分和梯度程序。
-
Cerius2系列的图形用户界面C2MOLCAS,可以简化输入,显示计算的MO,频率和结构。
-
到Molden的接口,用于绘制分子轨道,显示几何和谐振分析的结果。
-
格点显示程序MOLCASGV,基于OpenGL/Mesa,用于显示MOLCAS的分子的轨道、密度和密度差。
-
用GRID_IT包计算分子轨道和密度格点文件,用于MOLCASGV,C2MOLCAS和GABEDIT显示。
-
了MOPAC,HONDO等的功能。
-
到LUCIA和LUCIAREL的接口(需要向作者索取LUCITA程序),分别进行direct-CI和相对论双值群CI计算。


- 2026-07-24
- 2026-07-23
- 2026-07-21
- 2026-07-15
- 2026-07-15
- 2026-07-15

- 2026-07-24
- 2026-07-24
- 2026-07-24
- 2026-07-23
- 2026-07-23
- 2026-07-23










